ABSTRACT
Hepatocellular adenoma (HCA) is a rare benign liver tumor in the pediatric population, often associated with underlying genetic syndromes or metabolic disorders. In prepubertal children, the most common subtypes are β-catenin–mutated HCA and HNF1α-inactivated HCA (H-HCA), each with distinct clinical and prognostic implications. Differentiating HCA from malignant lesions such as hepatocellular carcinoma (HCC) is critical for appropriate management. We report a 2-year-old girl with hepatic failure, who was found to have multiple hepatic nodules. Imaging studies suggested features highly indicative of HCC. Due to multiple suspicious lesions and severe liver dysfunction, liver transplantation (LT) was performed. Histopathological examination confirmed the lesions as H-HCAs. The patient was postoperatively diagnosed with hereditary tyrosinemia type 1. This case highlights the importance of including HCA in the differential diagnosis of hepatic nodules in children with metabolic disorders. Accurate diagnosis of HCA is essential to guide clinical decision-making, optimize treatment strategies including LT.
-
KEYWORDS: Adenoma, liver cell; Hepatic adenomatosis; Tyrosinemia; Hepatic failure; Liver transplantation
INTRODUCTION
Hepatocellular adenoma (HCA) is a rare benign liver tumor in the pediatric population, with a significantly lower incidence compared to adults. While adult HCAs are most often sporadic and associated with risk factors such as oral contraceptive use or anabolic steroid exposure, pediatric HCAs typically arise in the context of underlying genetic syndromes or metabolic disorders. This association is particularly prominent in prepubertal children, in whom most reported HCAs are linked to systemic conditions.
Advances in immunohistochemical staining have enabled the subclassification of HCA into distinct molecular subtypes, each with differing clinical implications and prognostic relevance. In prepubertal children, the most frequently encountered subtypes are β-catenin–mutated HCA and HNF1α-inac-tivated HCA (H-HCA).
In this report, we describe the case of a 2-year-old girl with hereditary tyrosinemia type 1 (HT-1) complicated by hepatic failure, in whom multiple HCAs were identified. These lesions demonstrated imaging features highly suggestive of hepatocellular carcinoma (HCC). Because multiple tumors suspicious HCC and hepatic failure, liver transplantation (LT) was the treatment choice in this case. Through a detailed review of the clinical, pathological, and diagnostic findings, we aim highlight the importance of considering HCA in the differential diagnosis of hepatic nodules in pediatric patients with metabolic disorders.
CASE
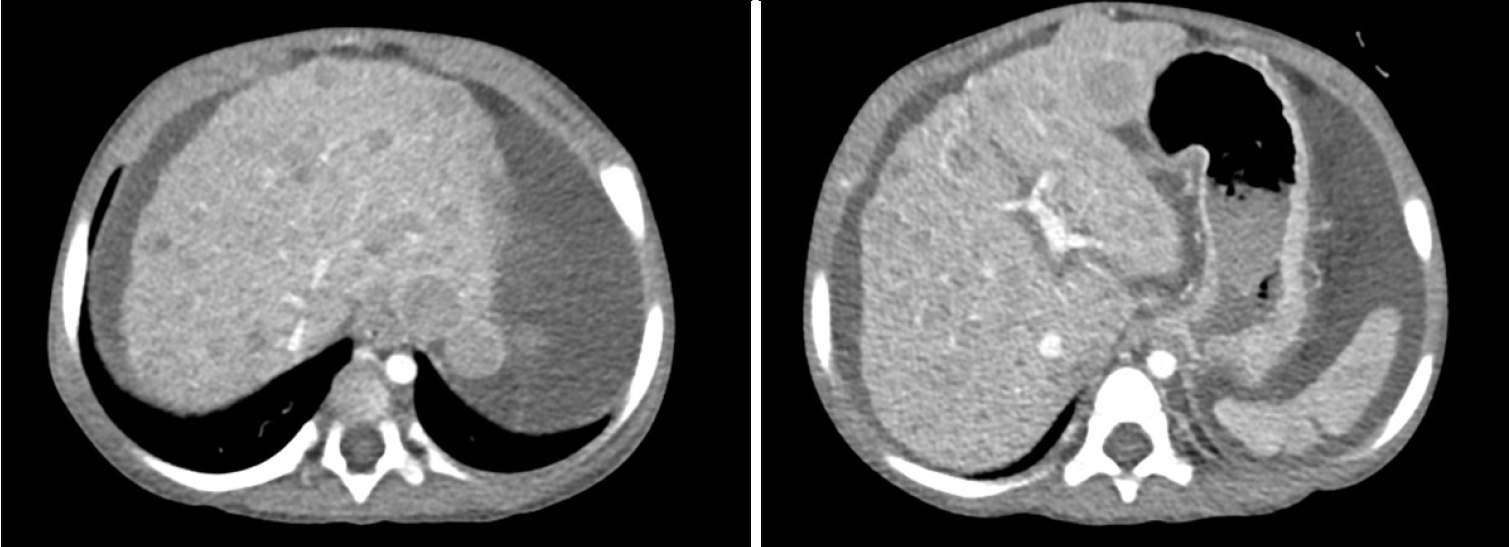
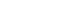
2-year-old child presented with jaundice and abdominal distension for 2 weeks. According to the patient’s parents, the patient had no known underlying medical history. Laboratory findings revealed hyperbilirubinemia, hypoalbuminemia, hyponatremia and coagulopathy. The findings were suggestive of impaired liver function. Renal function was unremarkable. Imaging studies, including computed tomography (CT) scan (
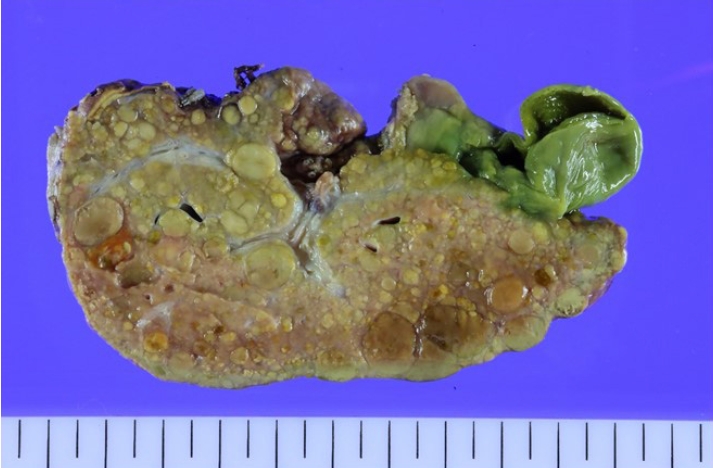
Fig. 1) and positron emission tomography CT, showed hepatomegaly with massive ascites and multiple tumors in whole liver, suspicious for HCC and alpha-fetoprotein (AFP) level was significantly elevated. There was no evidence of distant metastasis. Additionally, evaluation for suspected genetic disorders was performed in collaboration with the department of pediatrics, as the patient appeared to have delayed language development and motor impairment. Although the Pediatric End-Stage Liver Disease score was low (7), the patient’s condition was complicated by intractable massive ascites. Furthermore, as preoperative imaging and labs were strongly suggestive of malignancy, the patient’s parents agreed to proceed with liver transplantation. Her mother volunteered as a living donor and laparoscopic left lateral sectionectomy was performed for graft procurement. Postoperative course was uneventful. The donor discharged at postoperative day 7 without peri-operative complication, and the patient discharged at postoperative day 17. As the patient’s postoperative condition improved, language development and motor impairment showed normal age-ap-propriate progression. The patient has been under regular follow-up for three years without complication. In histopathologic examination, adenomatosis (more than 10 adenomas), suggestive of hepatocyte nuclear factor 1 homeobox alpha (HNF1a)-inactivated HCA was diagnosed. Focal dysplastic change and liver cirrhosis was also diagnosed. Multiple nodules were found in the specimen. (
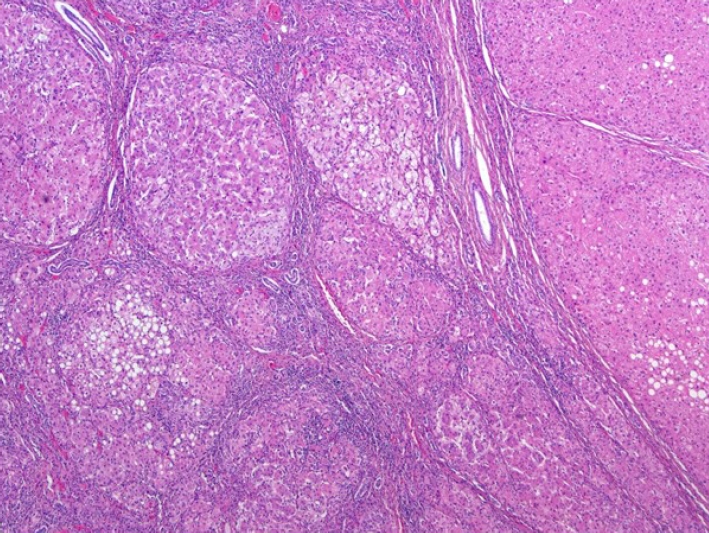
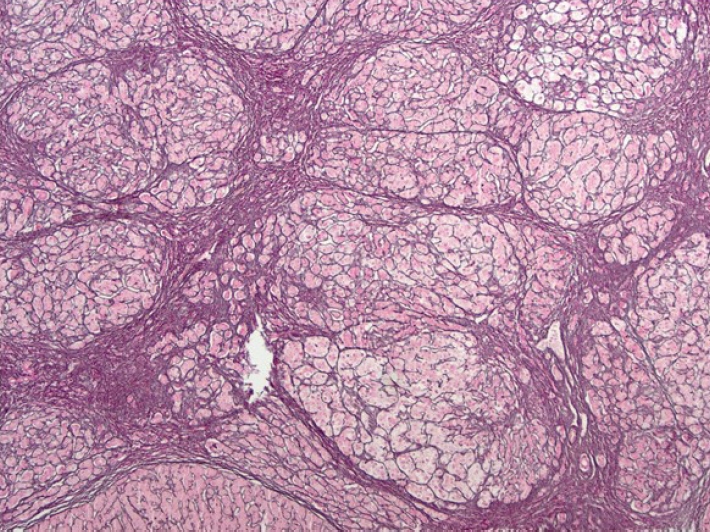
Fig. 2) There was presence of steatosis in tumor cell on Hematoxylin and Eosin staining. (
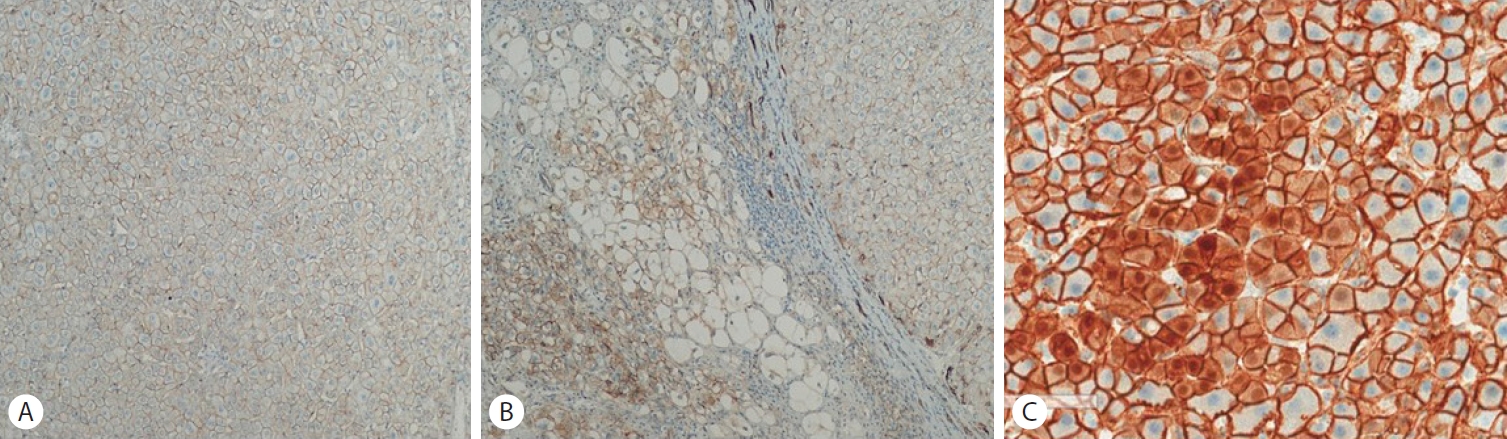
Fig. 3) In immunohistochemical staining, nuclear expression of beta catenin is characteristic of another subtype of HCA, but the tumor cells are negative for beta-catenin, (
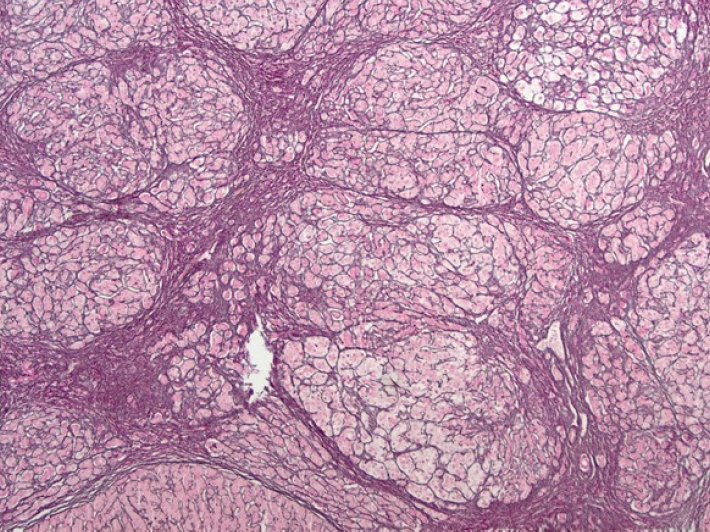
Fig. 4) the pathologist diagnosed HNF1a-inactivated HCA. The special staining was performed for reticulin. In reticulin staining (
Fig. 5), preserved pericellular pattern of reticulin framework and complete circling of small group of hepatocytes by reticulin fibers were found, it was the feature of HNF1a-inactivated HCA. Evaluation for genetic disorders revealed HT-1. Genetic screening of the family members was unremarkable, showing no evidence of hereditary metabolic diseases or predisposing genetic risk factors.
DISCUSSION
H-HCA accounts for approximately 35% of all HCAs [
1]. In the pediatric population, however, HCAs are rare and represent only 2.6% of primary liver tumors [
2]. H-HCA is typically characterized by diffuse steatosis and loss of liver fatty acid–binding protein expression on immunohistochemical staining, due to inactivating mutations of the HNF1α gene [
3]. In our patient, the histopathological features and immunohistochemical profile were consistent with H-HCA. Although malignant transformation of H-HCA has been reported to be extremely rare, ongoing surveillance is recommended to enable early detection of potential tumor progression.
Most HCAs are asymptomatic and are incidentally diagnosed. In this case, however, the chief complaints were symptoms associated with hepatic failure, and imaging studies revealed multiple hepatic masses. The differential diagnosis included the most common malignant liver tumor in children, such as hepatoblastoma and HCC. Hepatoblastoma is the most common malignant liver tumor in children and typically occurs in infants and young children younger than 3 years of age. It is frequently associated with markedly elevated serum AFP levels and has been linked to several congenital and genetic conditions, including Beckwith–Wiedemann syndrome and familial adenomatous polyposis. Hepatoblastoma usually presents as a solitary mass. Advances in multimodal treatment, including surgical resection and chemotherapy, have significantly improved survival outcomes. Pediatric HCC is the second most common malignant liver tumor in children and generally affects older children and adolescents. Unlike hepatoblastoma, it often arises in the setting of underlying chronic liver disease, such as biliary atresia or metabolic liver disorders. In rare cases, pediatric HCC may present with multiple hepatic masses and rapid deterioration of liver function, making early diagnosis and management challenging. Serum AFP levels may be normal or only mildly elevated, and the prognosis remains relatively poor compared with hepatoblastoma due to limited responsiveness to chemotherapy. In pediatric patients, a marked elevation of AFP typically raises immediate suspicion of a malignant germ cell tumor or hepatic tumor. However, HT-1 is a critical differential diagnosis, as it can present with both extreme AFP elevation and regenerative nodules that mimic the radiological appearance of a hepatic neoplasm [
4,
5]. While imaging may reveal multifocal nodules suggestive of malignancy, the nodules in HT-1 are often regenerative or dysplastic in nature. Although the initial marked elevation of AFP was suggestive of a primary liver malignancy, the final diagnosis was confirmed as HT-1. The histopathological finding of multiple adenomas with dysplastic nodules explains the diagnostic complexity of this case. Ultimately, it was concluded that the preoperative AFP surge was secondary to the underlying tyrosinemia, with the dysplastic changes representing the hepatic manifestation of the metabolic disorder rather than a discrete malignant tumor. Because portal hypertension was suspected, invasive procedures such as liver biopsy could not be performed, which made accurate preoperative evaluation difficult. Consequently, the parents consented to proceed with liver transplantation, and the diagnosis of H-HCA was confirmed postoperatively.
Several inherited and metabolic disorders have been implicated in the pathogenesis of pediatric HCA [
6]. Among these, glycogen storage disease is the most well-documented predisposing condition. Vascular anomalies and other metabolic diseases, such as HT-1, have also been associated with the development of hepatic adenomas, although reports remain relatively limited. In our patient, HT-1 was genetically confirmed postoperatively. HT-1 is an autosomal recessive disorder caused by deficiency of fumarylacetoacetate hydroxylase and presents with heterogeneous clinical manifestations. If untreated, patients with HT-1 may develop HCC [
7]. In cases of liver failure, including HCC or suspected HCC, LT is considered the treatment of choice. However, the occurrence of multiple HCAs in HT-1 is unusual and may radiologically and clinically mimic HCC, creating diagnostic and therapeutic challenges. In the present case, the indications for LT were liver failure secondary to HT-1 and suspected HCC [
8,
9].
In summary, this case highlights H-HCA with unique genetic and pathological characteristics in the context of HT-1. LT was performed urgently due to clinical manifestation of liver cirrhosis. Examination of the explanted liver in this pediatric patient provided valuable insights into liver adenomatosis. Pediatric hepatocellular lesions share many features with those in adults, but some distinctions exist [
10]. Recent advances in molecular diagnostics have improved the understanding of various pediatric hepatocellular neoplasms and may be similarly applied to adult cases. Ultimately, awareness of the histomorphology, immunohistochemistry, and genetic alterations associated with hepatic neoplasms is essential for distinguishing between entities and elucidating tumor biology. Further investigation is warranted to establish individualized treatment strategies tailored to each disease entity.
NOTES
-
ACKNOWLEDGEMENTS
We express our gratitude to the surgical, nursing, pathology, and genetic laboratory teams for their invaluable contributions to the patient’s clinical care, histopathologic evaluation, and genetic diagnosis.
-
FUND
None.
-
ETHICS STATEMENT
Consent for publication is not required, as this submission does not include any images or information that could identify any individual.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
JaRyung Han, Young Seok Han conceptualized manuscrpt, collected data, wrote the manuscrpt. Young Seok Han critically revised the manuscript.
Figure 1.The initial computed tomography scan showed multiple liver masses throughout the liver and massive ascites.
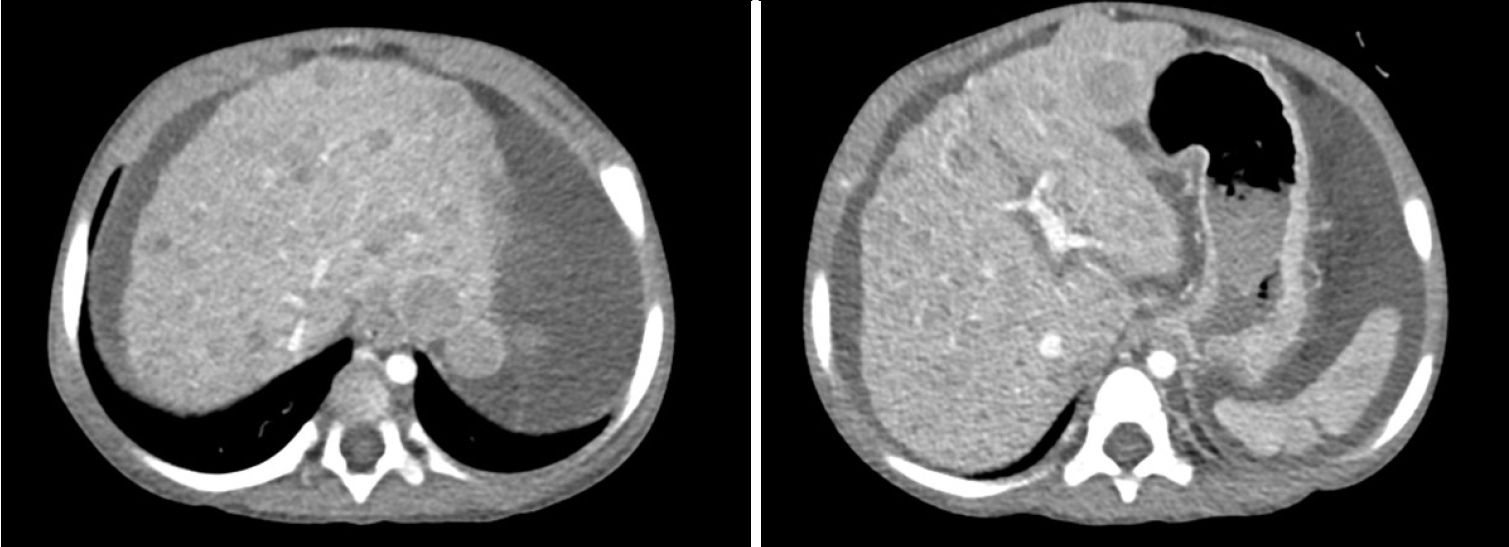
Figure 2.Gross examination of the explanted liver revealed multiple nodules.
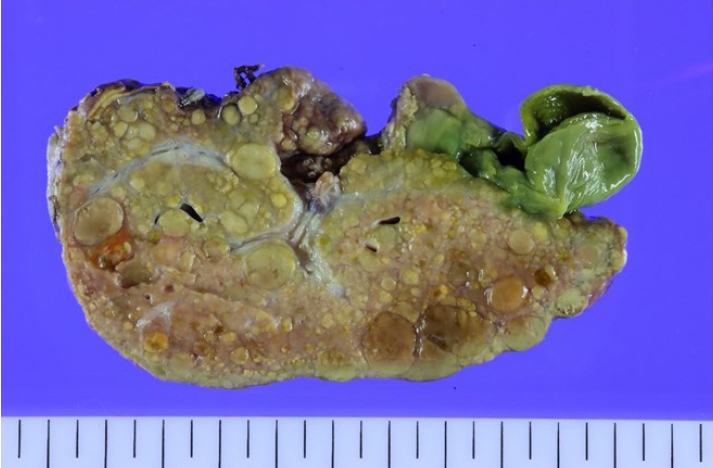
Figure 3.Steatosis was observed in the tumor cell on H&E staining (x40).
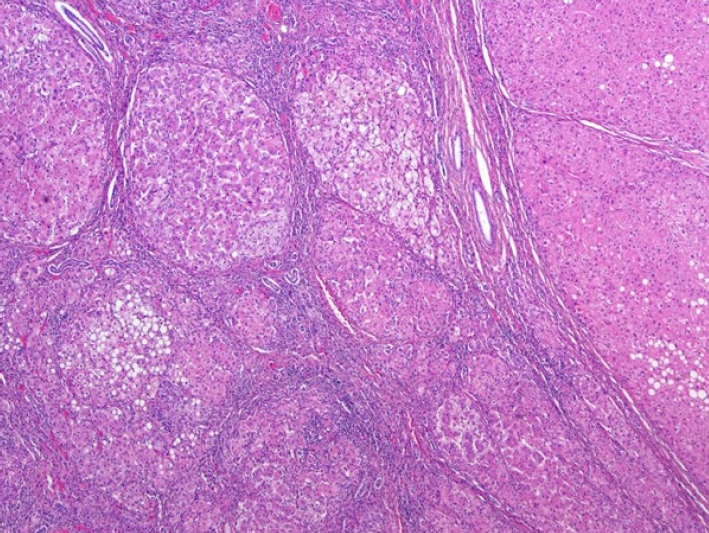
Figure 4.(A, B) Beta-catenin is not expressed in the specimen (Beta-catenin staining, x40). (C) Nuclear expression of beta catenin is characteristic of another subtype of HCA (x20). HCA, hepatocellular adenoma.
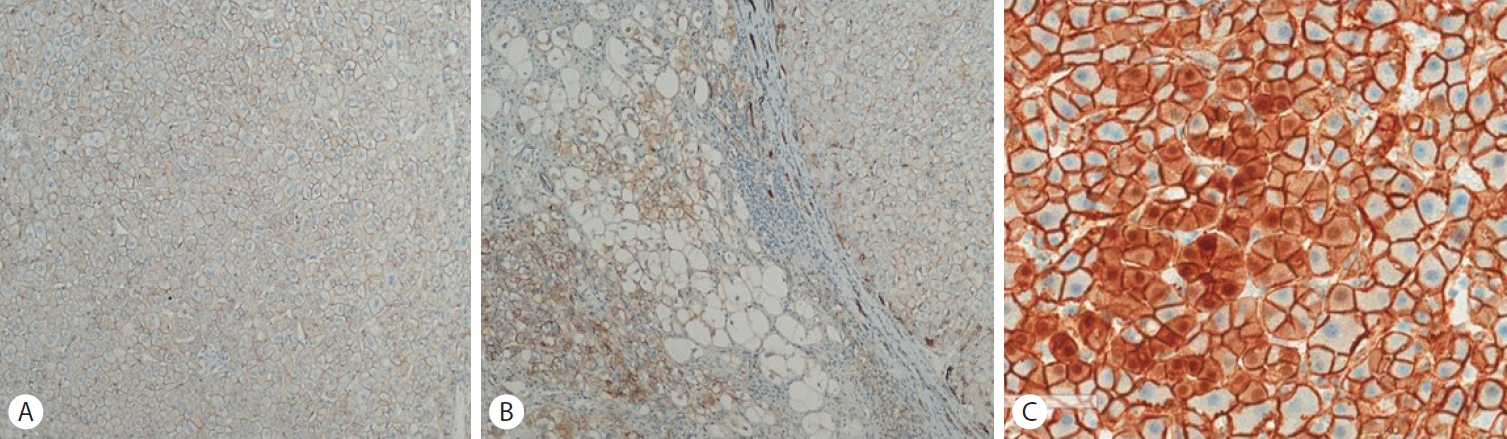
Figure 5.The typical features of HNF1α-inactivated HCA are seen on reticulin staining (x40). HCA, hepatocellular adenoma.
REFERENCES
- 1. Nault JC, Paradis V, Cherqui D, Vilgrain V, Zucman-Rossi J. Molecular classification of hepatocellular adenoma in clinical practice. J Hepatol 2017;67:1074-1083.
- 2. Muthuvel E, Chander V, Srinivasan C. A clinicopathological study of paediatric liver tumours in a tertiary care hospital. J Clin Diagn Res 2017;11:EC50-EC53.
- 3. Jeannot E, Mellottee L, Bioulac-Sage P, et al. Spectrum of HNF1A somatic mutations in hepatocellular adenoma differs from that in patients with MODY3 and suggests genotoxic damage. Diabetes 2010;59:1836-1844.
- 4. de Laet C, Dionisi-Vici C, Leonard JV, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 2013;8:8.
- 5. Joshi SN, Venugopalan P. Experience with NTBC therapy in hereditary tyrosinaemia type I: an alternative to liver transplantation. Ann Trop Paediatr 2004;24:259-265.
- 6. González IA, Pacheco MC. What is new in pediatric hepatic neoplasms. Surg Pathol Clin 2025;18:281-300.
- 7. Hepkema JT, Poelmann FB, Gouw ASH, et al. Malignant transformation of an HNF1a-inactivated hepatocellular adenoma to hepatocellular carcinoma. Case Rep Gastroenterol 2020;14:577-585.
- 8. Chiche L, David A, Adam R, et al. Liver transplantation for adenomatosis: European experience. Liver Transpl 2016;22:516-526.
- 9. Kawabata K, Kido J, Yoshida T, Matsumoto S, Nakamura K. A case report of two siblings with hypertyrosinemia type 1 presenting with hepatic disease with different onset time and severity. Mol Genet Metab Rep 2022;32:100892.
- 10. Pacheco MC, Torbenson MS, Wu TT, Kakar S, Jain D, Yeh MM. Pediatric hepatocellular adenomas: the influence of age and syndrome on subtype. Am J Surg Pathol 2021;45:1641-1647.
Citations
Citations to this article as recorded by